





Le séquençage des variants du Covid expliqué par ses spécialistes
Le « séquençage » du génome du SARS-CoV-2 est sur toutes les lèvres, pour repérer les variants, décrypter les conséquences des changements de sa protéine Spike…
Mais concrètement, de quoi s’agit-il ?
Le séquençage des variants du Covid expliqué par ses spécialistes
Fin 2019 débutait une épidémie due à l’émergence d’un nouveau virus, le SARS-CoV-2, qui allait causer une maladie inconnue, le Covid-19. Rapidement, le pathogène originel allait être remplacé par des mutants, des variants – Alpha, Delta… et Omicron qui domine aujourd’hui la scène internationale.
Comment identifie-t-on ces variants ? Comment les nomme-t-on ? Comment les classe-t-on ? Tout repose sur le « séquençage », la lecture du génome du virus : en l’occurrence un texte génétique d’environ 30 000 bases (ou « lettres »). C’est cette technique devenue incontournable et que nous mettons en œuvre quotidiennement au laboratoire de virologie du CHU de Rouen, que nous vous proposons de décrypter ici.
Pour commencer, quelques rappels de virologie. Le génome du SARS-CoV-2 contient les informations nécessaires à sa réplication et à la production de sa trentaine de protéines – dont les « briques », les acides aminés, sont assemblées d’après l’ordre des bases dans son génome.
Parmi ces protéines, la désormais célèbre protéine S permet au virus de se lier à sa cellule cible : elle joue donc un rôle majeur dans l’infection. Elle est aussi reconnue par les anticorps générés par notre système immunitaire, après maladie ou vaccination.
Depuis le début de l’épidémie, le génome du SARS-CoV-2 a changé : il a évolué du fait des nombreuses mutations qui s’y sont produites, sorte d’erreurs apparaissant lors de sa réplication. Elles peuvent être de différentes natures : substitution d’une base par une autre, insertion ou suppression d’une base (délétion)… ce qui entraîne un changement dans les acides aminés, et donc de la protéine associée.
Chaque mutation dans une protéine est identifiée par un code constitué d’une lettre désignant l’acide aminé avant mutation, sa position dans le texte génétique et la lettre de l’acide aminé après mutation. Ainsi, à la position 614 de la protéine S, le remplacement de l’acide aminé acide aspartique (codé D) par une glycine (codée G) s’écrit « D614G ».
Le suivi de l’évolution des mutations a permis de définir des clades et des lignages, correspondant à différents groupes de virus partageant les mêmes mutations.
Certaines mutations modifient les propriétés des virus, parfois au point d’avoir un impact sur l’épidémie elle-même : transmissibilité, tableau clinique et sévérité voire efficacité des outils diagnostiques ou des traitements et vaccins peuvent être affectés. Les virus concernés sont alors désignés comme variants. Ainsi, les mutations E484K (présentes notamment chez Bêta et Gamma), N501Y (chez Omicron, Alpha, Bêta et Gamma) et L452R (Delta) semblent liées à un échappement immunitaire et/ou à une augmentation de transmissibilité.
« Ranger » les variants
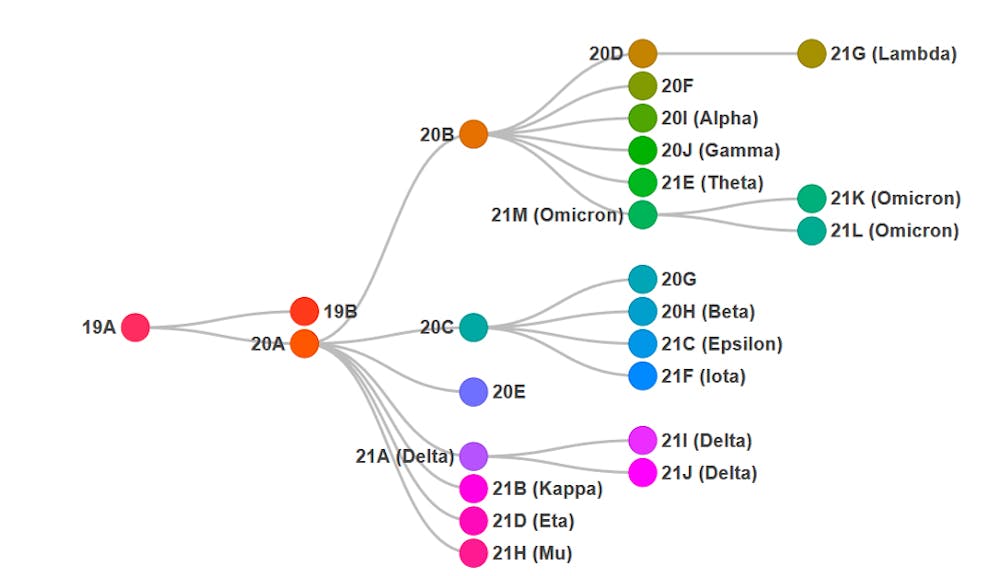
Pour s’y repérer et classer ces variants, deux modes de « rangement » (complémentaires mais qui ne se recouvrent pas) sont à disposition des scientifiques : les clades Nextstrain, identifiés par une année et une lettre (19, 20 ou 21 donnant par exemple 21K, 21L et 21M pour Omicron), et les lignages dits « Pangolin » (rien à voir avec l’animal), composés de lettres et de chiffres (BA.1, BA.2 et BA.3 toujours pour Omicron).
Ils correspondent, en gros, à des « familles » de virus. L’utilisation des clades et lignages (et d’une lettre grecque le cas échéant) permet d’assurer un consensus international quant à l’identité d’un variant.
 Illustration des relations phylogénétiques entre les différents clades (familles) du SARS-CoV-2,
Illustration des relations phylogénétiques entre les différents clades (familles) du SARS-CoV-2,
tels que définis par le projet international Nextstrain. clades.nextstrain.org
En fonction de l’impact potentiel d’un variant émergent sur la transmissibilité, la gravité, la présentation clinique et l’efficacité des mesures de contrôle et de prévention (outils diagnostiques, vaccination, molécules thérapeutiques), l’Organisation mondiale de la santé (OMS) le classe variant préoccupant (VOC), variant d’intérêt (VOI) ou variant à surveiller (VUM). Seuls les VOC sont associés à une lettre grecque. Ainsi, le variant Omicron regroupe plusieurs clades (21K/L/M) et plusieurs lignages (B.1.1.529 BA.1, BA.2 et BA.3).
En France, une analyse de risques sur les variants émergents du SARS-CoV-2 est réalisée conjointement par Santé publique France et les CNR (Centres nationaux de référence) des virus des infections respiratoires, tous les 15 jours.
Classement des trois types de variants du SARS-CoV-2 en France, d’après l’analyse des risques au 2 janvier 2022.
santepubliquefrance.fr/dossiers/coronavirus-covid-19/coronavirus-circulation-des-variants-du-sars-cov-2

Repérer les mutations
Nous entrons désormais dans le vif du sujet puisque la présence de mutations se détecte en laboratoire par séquençage. Des « séquenceurs » lisent le texte du génome d’un virus donné, ce qui permet par comparaison avec les séquences déjà connues, de noter les différences et donc repérer les variants et les analyser. On peut ainsi repérer la présence ou l’apparition de variants (anciens ou inédits), tracer leur diffusion dans l’espace et dans le temps et suivre l’évolution de l’épidémie.
Mais les données obtenues ne sont pas interprétables telles quelles et nécessitent une analyse bio-informatique. Les données brutes générées par le séquenceur doivent être traitées pour restituer une séquence virale lisible.
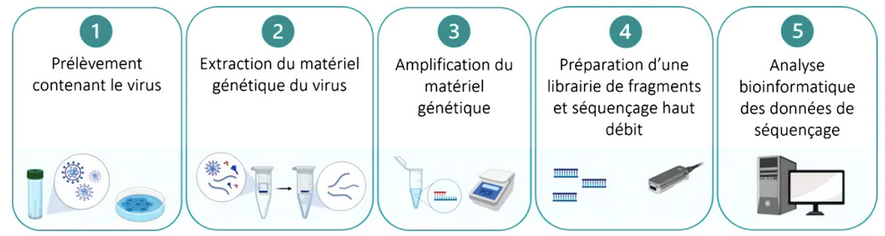 Étapes du séquençage haut débit d’un génome de SARS-CoV-2, du prélèvement sur un patient à l’analyse des résultats.
Étapes du séquençage haut débit d’un génome de SARS-CoV-2, du prélèvement sur un patient à l’analyse des résultats.
L’ensemble demande cinq jours. D’après Hourdel V. et col., Front. Microbiol. 11:571328–2020, Author provided (no reuse)
Lisant l’intégralité du texte génétique et pouvant donc repérer des variants inédits, le séquençage est particulièrement puissant… Mais cette technique reste longue : entre la préparation des échantillons, la « lecture » proprement dite dans l’automate, qui peut durer plusieurs dizaines d’heures, et le traitement des résultats jusqu’à validation, il se passe environ cinq jours. Malgré une amélioration de l’équipement, la plupart des laboratoires n’ont ainsi pas la capacité de séquencer la totalité des prélèvements positifs à SARS-CoV-2. Actuellement, en France, environ 1 à 2 % des cas de Covid-19 font l’objet d’un séquençage.
Pour le suivi au quotidien, les recommandations nationales imposent la réalisation d’une seconde RT-PCR, dite de criblage, sur la plupart des prélèvements positifs après la RT-PCR de dépistage. Le criblage permet de repérer des mutations précises, définies au préalable, et sert donc à traquer des variants déjà connus. Et le résultat est cette fois obtenu en 24 à 48 heures.
Les situations où le séquençage est nécessaire ont été définies en France à l’échelle nationale. Il s’agit notamment des cas positifs après retour de l’étranger, d’échecs de vaccination ou de traitements par anticorps monoclonaux ou des cas d’infections avec des excrétions virales prolongées, en particulier chez les personnes immunodéprimées. En parallèle, afin de surveiller la distribution et la circulation en temps réel des différents variants du SARS-CoV-2, un séquençage aléatoire est réalisé de manière hebdomadaire, dans le cadre des enquêtes FLASH# nationales.
La puissance du séquençage
Le séquençage prend tout son intérêt lorsque les jeux complets de données générées de chaque laboratoire (séquences génétiques, données cliniques et épidémiologiques) sont compilés et étudiés à grande échelle.
Cela permet par exemple de modéliser des outils diagnostiques, de traitement et des vaccins. Au niveau de l’épidémie, il permet aussi d’établir une cartographie, nationale ou internationale, des variants circulant à un instant donné, et de déceler précocement une émergence sur un ou plusieurs territoires ou dans une ou plusieurs populations.
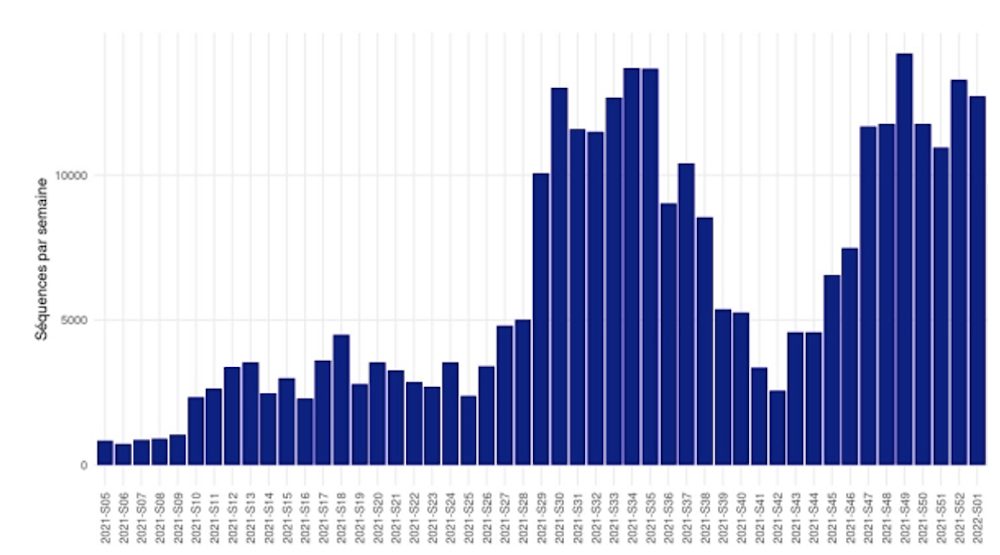
C’est dans cette optique qu’a été mis en place, en France, le consortium EMERGEN, coordonné par Santé publique France et l’ANRS-Maladies infectieuses émergentes (ANRS|MIE). Son objectif est d’assurer une surveillance génomique des infections à SARS-CoV-2. Une quarantaine de laboratoires du réseau ANRS-MIE sont impliqués. Depuis le 03 janvier 2021, 342 419 séquences au total ont été produites chiffres Santé Publique France au 17 janvier 2022, dont 2 117 provenant de notre laboratoire de Virologie, au CHU de Rouen.
Activité nationale de séquençage, basée sur le nombre de prélèvements reçus, semaines 2021-S5 à 2022-S1. Consortium EMERGEN
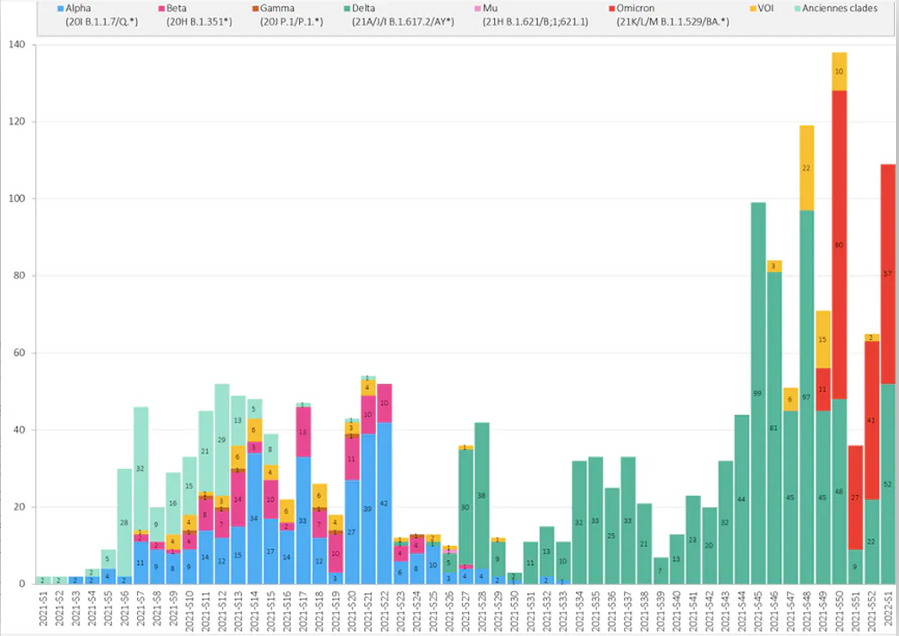
L’activité de séquençage et la répartition hebdomadaire des clades séquencés à Rouen, entre janvier 2021 et 2022, sont présentées ci-dessous. La proportion des différents clades doit être analysée en prenant en compte certains biais de sélection, puisque les recommandations de séquençage ont évolué au cours de l’année, conduisant à une surreprésentation de certains variants au détriment d’autres.
Répartition hebdomadaire des clades séquencés au CHU de Rouen, semaines 2021-S1 à 2022-S1 : on voit comment Omicron (rouge) prend le pas sur Delta (vert),
qui avait lui-même supplanté les autres variants. A. Moisan, F. De Oliveira, E. Alessandri-Gradt, J.-C. Plantier, Author provided (no reuse)
À l’échelle internationale, les données de séquençage du SARS-CoV-2 sont partagées, en temps réel, avec la communauté scientifique grâce à la base de données GISAID, disponible en libre accès. La France se place au 6e rang mondial des contributeurs sur GISAID, avec 193 655 séquences déposées depuis janvier 2020, et au 3e rang des pays de l’Union européenne, après l’Allemagne et le Danemark – comblant ainsi le retard précédemment constaté.
L’étude de la proportion des différents clades, tant au niveau local que national et international, montre un ralentissement de la diversification de l’épidémie avec de moins en moins de variants co-circulant sur un même territoire et à un même moment.
Circulation des clades de SARS-CoV-2 dans le monde, entre février 2021 et janvier 2022
(en rouge, Omicron ; en vert, Delta ; en violet, Alpha). nextstrain.org/ncov/gisaid/global

Ainsi, après cinq mois de circulation quasi exclusive du variant Delta en France, le variant Omicron a émergé rapidement à la fin de l’année 2021 et représentait 95.6 % des virus séquencés au 10 janvier 2022.
Ce variant est associé à une transmissibilité plus élevée mais à un risque d’hospitalisation et de sévérité plus faibles que pour Delta (chez les vaccinés trois doses). Ces caractéristiques lui permettent d’avoir une meilleure diffusion dans la population.
S’il n’est pas évident à repérer, il reste identifiable par criblage selon les combinaisons de mutations ciblées par la technique. Ainsi, certaines délétions dans la protéine Spike présentes chez BA.1 mais pas chez BA.2 offrent de premiers indices qu’un séquençage complet peut confirmer. Début février 2022, le lignage BA.2 représentait entre 1 à 3 % des variants circulant sur le territoire français.
Le déploiement important du séquençage haut débit, tant à l’échelle nationale qu’internationale, permet désormais de repérer rapidement l’émergence d’un variant. Cela constitue un pilier majeur de lutte contre la pandémie puisqu’il permet une adaptation immédiate des stratégies de prévention : en préconisant de nouvelles doses vaccinales si les premières données suggèrent un échappement immunitaire, mise en place de jauges, confinement, télétravail…
De manière intéressante, le séquençage met actuellement en évidence un ralentissement de la diversification du SARS-CoV-2 : avec désormais une souche majoritaire écrasant tous les anciens clades ayant circulé depuis le début de l’épidémie il y a deux ans.
Auteurs
Alice Moisan, AHU en virologie, Université de Rouen Normandie
Elodie Alessandri-Gradt, MCU-PH en virologie, CHU de Rouen, Université de Rouen Normandie
Fabienne De Oliveira, Ingénieure Hospitalier, Université de Rouen Normandie
Jean-Christophe Plantier, Professeur des Universités – Praticien Hospitalier en Virologie, Université de Rouen Normandie
Cet article est republié à partir de The Conversation sous licence Creative Commons. Lire l’article original.
Dernière mise à jour : 27/04/22
Date de publication : 16/02/22